
PCR Protocol
PCR Protocol for POC and POR Species ID (Can be modified for other primers)
See base protocol and notes from Hollie. This protocol format is based off of Maggie’s, with adjustments to the protocol in advisement from Kristina (notable differences: no diluting DNA beforehand and strip tubes instead of plates).
Goal
Amplify the mtORF region (mitochondrial open reading frame) of Pocillopora and Porites coral DNA using PCR (polymerase chain reaction). Then use amplification product for Sanger Sequencing and determination of Pocillopora coral species. Primer sequences and concept developed in Johnston et. al 2018 and Tisthammer et al 2020.
Information on Sanger sequencing at the URI GSC is here. Sequencing takes place after 10am on Tuesdays and Thursdays, so samples need to be dropped of before 10am on those days.
Materials and Equipment
- Primers (orderable from IDT), see specific primers for each species below, stored in -20 ºC
- EmeraldAmp® GT PCR Master Mix Cat # RR310A RR310B, stored in -20 ºC
- Ultrapure free water/PCR grade water, stored in -20 ºC
- Thermocyler
Pocillopora
mtORF: Johnston et al. 2018
mtORF Primers
FatP6.1 5′-TTTGGGSATTCGTTTAGCAG-3′
RORF 5′-SCCAATATGTTAAACASCATGTCA-3′
mtORF PCR profile
1 cycle: 94 °C for 60 s
30-40 cycles: 94 °C for 30 s, 53 °C for 30 s, and 72 °C for 75 s
1 cycle: 72 °C for 5 min.
PocHistone 3 region: Johnston et al. 2018
DNA from individuals of mtORF type 1 (P. meandrina and P. eydouxi) need also to be amplified using novel primers for the histone 3 region
PocHistone Primers
PocHistoneF: 5′-ATTCAGTCTCACTCACTCACTCAC-3′ PocHistoneR: 5′-TATCTTCGAACAGACCCACCAAAT-3′
PocHistone PCR profile:
1 cycle: 94 °C for 60 s
30-40 cycles: 94 °C for 30 s, 53 °C for 30 s, and 72 °C for 60 s
1 cycle: 72 °C for 5 min.
Porites
Coral nuclear histone region spanning H2A to H4 (H2)
Reference: Tisthammer et al. 2020
H2 Primers
zH2AH4f 5′-GTGTACTTGGCTGCYGTRCT-3′
zH4Fr 5′-GACAACCGAGAATGTCCGGT-3′
H2 PCR profile
1 cycle: 96 °C for 2 min
34 cycles: 96 °C for 20 s, 58.5 °C for 20 s, and 72 °C for 90 s
1 cycle: 72 °C for 5 min.
expected band size ∼1,500 bp
Protocol Steps
- Primer working stock dilution
- PCR amplification
- 1% Gel to check controls and amplicon length
- Ethanol precipitation
- Nanodrop or Qubit
- DNA dilution and prep for sequencing
All references to H2O refer to PCR-grade, nucelic acid, DNAse, and RNAse free H2O.
Primer Hydration from shipped dried primers (to 200µM)
IDT’s website has a link to a “Resuspension Calculator” that you can use to hydrate the primers to a concentration of 200µM. You will need to make an account to use the calculator, and you tell it the concentration you want to dilute to (200µM) and the “OD” number in units “nmol” written on the tube of the primer, which will be different for each tube. For example, for primer zH2AH4f, the OD # was 93.5 nmol and I wanted a final concentration of 200µM, so the calculator told me to add 467.5 µL of Low TE buffer or PCR-grade H2O.
Make sure to spin down the tube with the dehydrated primers before opening it so that none escapes!
Primer Dilution to 10 µM from Hydrated Primers (200 µM)
Then, make a stock solution of the primers for yourself for daily use. This will have a concentration of 10 µM:
25 µL of 200 µM primer + 475 µL PCR-grade H2O
Store in -20 ºC freezer with other PCR reagents (H2O, MasterMix)
PCR Amplification
For all primer sets below, I used the same reaction formula:
| Master Mix 25 µL reaction | |
|---|---|
| 12.55 | Emerald Master Mix |
| 10.80 | Nuclease Free Water |
| 0.32 | 10µM working stock of For Primer |
| 0.32 | 10µM working stock of Rev Primer |
| 1.00 | DNA Template |
| 25 | Total Volume |
This is based on a 33 µL formula from Kristina:
| Master Mix 33 µL reaction | |
|---|---|
| 16.66 | Emerald Master Mix |
| 14.66 | Nuclease Free Water |
| 0.43 | 10µM working stock of For Primer |
| 0.43 | 10µM working stock of Rev Primer |
| 1 | DNA Template |
| 33.18 | Total Volume |
For each amplification, you will need to have a positive control and a negative control. Ideally, the positive control is a sample of DNA that has been successfully amplified using that primer set before. If you don’t have this, you can probably skip the positive control. For the negative control, add everything except for the DNA into the reaction. The negative control is the most important because it will show a sign of any contamination in your reaction, primers, master mix, or water.
- Each sample will be amplified one time in a 25µL reaction volume
- Calculate the amount of master mix is needed: add number of samples and controls plus a % error (5-10%) to get the n number (ex: for 15 samples, make enough for n = 18)
- Make master mix in 1.5mL or 5mL tube with these components scaled up to your n number:
- 12.55µL Emerald PCR master mix * n =
- 10.80µL UltraPure water * n =
- 0.32µL 10µM working stock FatP6.1 primer * n =
- 0.32µL 10µM working stock RORF primer * n =
- add water first, then primers, then Master Mix, always load least expensive reagent –> most expensive
- Vortex and spin down master mix and keep on ice.
- Into labeled PCR strip tubes, add 24µL of master mix to as many tubes as samples you have + the number of positive and negative controls planned. Positive and negative controls could be in a separate strip tube or individual 0.2 mL tubes since they do not need to be kept after the gel imaging step.
- Using a p2 pipette, add 1µL of DNA sample to their respective tube where the master mix is added. Add in 1 µL of H2O (or nothing) to the tube for the negative control and add 1 µL of the positive control DNA to the positive control tube. You should have 25µL in each tube.
- Spin down in microcentrifuge with strip-tube attachment.
- Turn on thermocyler and login to PUTNAM (1,2,3,4) and navigate to the program (in Sanger folder) for the given primer set.
- POC mtORF program: FATP RORF, bold fields are cycled 30 times
- 60 seconds 94 ºC
- 30 seconds 94 ºC
- 30 seconds 53 ºC
- 75 seconds 72 ºC
- 5 minutes 72 ºC
- POC PocHistone program: PocHistone, bold fields are cycled 30 times
- 60 seconds 94 ºC
- 30 seconds 94 ºC
- 30 seconds 53 ºC
- 60 seconds 72 ºC
- 5 minutes 72 ºC
- POR H2 program: POC zH2AH4f, bold fields are cycled 34 times
- 120 seconds 96 ºC
- 20 seconds 96 ºC
- 20 seconds 58.5 ºC
- 90 seconds 72 ºC
- 5 minutes 72 ºC
- POC mtORF program: FATP RORF, bold fields are cycled 30 times
- Run that program, bold fields are cycled 30-40 times depending on the program (see details above).
- The program runs for about 1 hour and 40 minutes
- Take tubes out and store at 4 ºC if not running gel / cleaning up immediately
DNA and RNA Quality Check: 1% Gel at 80V for 30 mins.
Dilute 1kb ladder: 6 µL H2O, 2 µL of ladder, and 2 µL of loading dye, load 5 µL of this mixture per row.
Load 4 µL of the 25 µL of PCR product into the gel.
No loading dye needed to add to samples because of the green dye in the Master Mix. Use a 1kb DNA ladder. For POC mtORF, there should be 1 band at ~1000bp. For POR H2, there should be one band at ~1500bp but the paper (see above) has also reported cases of double banding, so if you see multiple bands for this primer set it will take some digging into the literature and possibly using other primer sets for that sample (see base protocol and notes from Hollie for notes about other Porites primers sets, many of which we already have in the lab). Other bands are signs of potential contamination or off target amplification.
Ethanol precipitation to clean up PCR product
- Add 2.1µL of 3M sodium acetate to each PCR reaction (1/10th the total voume)
- Add 69.3µL of ice cold 100% ethanol to each tube (3x the total volume after adding sodium acetate)
- Pipette up and down to mix and transfer entire volume to a new labeled 1.5ml tube
- Place tubes in -80 ºC freezer for 1 hour or in the -20 ºC freezer overnight
- Spin for 30 minutes at 15,000rcf (room temp). Make sure all the tubes are facing the same way (hinges out). The DNA will pellet on the back wall
- Pipette off the liquid without disturbing the pellet. It will most likely be invisible.
- Add 200µL of ice cold 70% Ethanol
- Spin for 15 minutes at 15,000rcf. This is to wash the pellet. Again make sure each tube is facing the same direction
- Pipette off the ethanol without disturbing the pellet
- Let dry at room temperature with the caps open for about 15 minutes or until all ethanol has evaporated
- Add 30µL of Tris to the pellet and pipette up and down to resuspend, go straight to nanodrop/qubit quantification or store cleaned PCR product in the -20 ºC freezer.
Nanodrop or Qubit of cleaned PCR product
Two options:
-
Follow Broad Range dsDNA Qubit protocol to analyze cleaned PCR product. Read standards once and record values, read all samples twice.
-
Nanodrop the samples:
- The standard PPP Nanodrop protocol for RNA can be found here.
- I used the nanodrop in the Lane lab with supervision/permission from Kristina Terpis, the Technician in the Putnam and Lane labs. The protocol is the same but it only can read one sample at a time and there is no printer, so I record the values by hand and take a picture of the screen.
- Blank nanodrop with the same TRIS used to elute the cleaned PCR product.
For Sanger Sequencing
Primer Dilution from 10 µM Primer Working stock to 3.2 µM for sequencing
1.6 µL of 10 µM primer + 3.4 µL H2O = 5 µL
For each primer that you are using to sequence (forward and reverse) make enough for # of samples times two so you have 2µL per reaction (e.g. 30 µL plus some extra for error)
Prep for sequencing (post PCR bench)
Notes
GSC Template to fill out, does the below calculations for you
- For Sanger sequencing at the URI GSC the amount of DNA to send them depends on how long the fragment is. The equation for how many ng to send is ((number of bases / 100)) * 1.25) * 2
- For mtORF the calculation is: ((1000/100)* 1.25) * 2 = 25. That means you’ll need 25ng of each amplification
- For PocHistone the calculation is: ((675/100)* 1.25) * 2 = 16.875. That means you’ll need 16.875ng of each amplification
- For H2 the calculation is: ((1500/100)* 1.25) * 2 = 37.5. That means you’ll need 37.5ng of each amplification
- 25 ng is not a lot of DNA, so some of your Qubit values are likely going to be higher than 25ng/µL. It’s best practice not to pipette below 1µL, so it’s probably you’ll need to dilute the amplifications.
- The volume of liquid sent to the GSC is 12µL total, and 2µL of that is one of the primers. So you have 10µL to use for DNA. Depending on the concentrations, you’ll have to decide if a 1:10, 1:5, 1:2, etc dilution will work to pipette over 1µL for 25 ng but under 10µL.
Tube labelling for GSC:
“Please identify your samples using the following code: Your initials followed by the number 1 to 9999 (i.e.: PJ1, PJ2, etc.). You should increment the number with each submission. This code facilitates instrument plate setup and data file management.” This is how Kristina taught me to label, and I would continue that pattern for #s 9-XX with the ZD on the first and last tube/lid as shown:
Steps for sequencing prep:
- Dilute DNA into new strip tubes. As an example, fill all the strip tubes (for appropriate # of samples x 2 for the forward and reverse primers) with 9µL of water and then add 1µL from each sample into the appropriately labelled tube. That would be a 1:10 dilution
- Dilute each of the primers to 3.2µM in volume for 2µL per sample (see above). Sanger sequencing amplifies the fragment with one of the two primers at a time, so you will submit two tubes per sample, one prepared with the forward primer and one prepared with the reverse primer.
- Calculate volume of DNA for each sample (use the dilution factor you used on your previous concentration values)
- Aliquot labelled strip tubes with the correct amount of DNA per sample and nuclease free water up to 10µL for each well (add water first, then DNA, then primers, always load least expensive reagent –> most expensive). Using a sample list here where you highlight off each well after adding the right amount is very helpful. For setting up forward and reverse reactions, each sample will be pipetted into two wells (i.e. tubes 1 & 2 will both contain sample POC1, tubes 3 & 4 will both contain sample POC2, so be very careful in this pipetting step because it can get confusing).
- example: 2.9 µL of 1:10 diluted DNA and 7.1 µL of H2O
- Add 2µL of 3.2µM FORWARD primer to the first tube for each sample (i.e. in the example above tubes 1 & 3) and 2µL of 3.2µM REVERSE primer to the second tube for each sample (i.e. in the example above tubes 2 & 4).
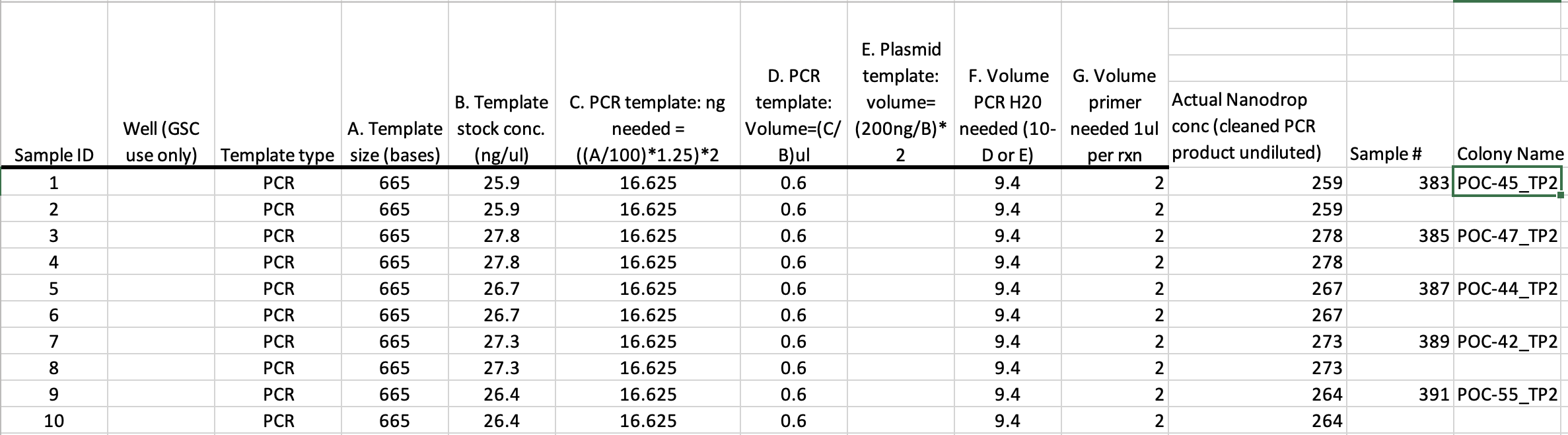
- Fill out the template linked above and print out two copies. One is for your records and can contain extraneous information such as your sample # (POC1 as opposed to the # given to GSC, ZD1), the undiluted concentration, and anything else you want to include in your lab notebook. The copy you print for GSC should have this info removed before printing. Either print this and bring it with you and add to the clipboard on the sample drop-off freezer or email this sheet to the GSC at jatoyan@uri.edu or gsc@etal.uri.edu saying you are submitting how ever many samples for Sanger sequencing for the Putnam Lab using her blanket PO (purchase order).
My copy:
GSC copy:
- Take plate/tubes up to the GSC and place in freezer next to the door. There is a place on the top shelf that with a label that says “samples for GSC sequencing”, place your tubes in that rack (right now it is purple).
You should get the sequences back the next day!
For RFLP Using Restriction Enzymes
After PocHistone amplification of a subset of mtORF samples that were amplified and identified to be mtORF type 1 (P. meandrina and P. eydouxi), we can do RFLP using the XhoI RE to try to differentiate the species. After checking the PCR product on the gel, send for sequencing as described above or do the following:
- Pipette 15ul of the PocHistone PCR product into a new labeled 0.2ml strip tube
- Add 0.5ul of 1x Cutsmart buffer (dilute from 10x tube) and 0.5ul of XhoI RE
- Incubated at 37 ºC for one hour, and then at 65 ºC for 20 minutes to inactivate the enzyme.
- Run a 2% gel for 1 hour at 70V (load 3ul from each tube)
- Analyze banding pattern on gel.
“DNA from individuals of mtORF type 1 (P. meandrina and P. eydouxi) were amplified using novel primers for this histone 3 region (PocHistoneF: 5′-ATTCAGTCTCACTCACTCACTCAC-3′ and PocHistoneR: 5′-TATCTTCGAACAGACCCACCAAAT-3′; accession numbers: MG587096–MG587097) (Table 1). PCR mixes were prepared as described above, and the following thermocycling protocol was used: denaturation for 60 s at 94 °C, followed by 40 cycles of 30 s at 94 °C, 30 s at 53 °C, and 60 s at 72 °C, with a final elongation step of 5 min at 72 °C. PCR products were digested with 0.5 μL of XhoI restriction enzyme (New England BioLabs, Ipswich, MA, USA) and 0.5 μL of 1X CutSmart® buffer for 1 hr at 37 °C, followed by heat inactivation at 65 °C for 20 min (Table 1). Three microliters of the digested products were run on a 2% TAE-agarose gel for 1.5 h at 70 V.”