
LCM Pico Methyl-Seq Library Prep
LCM Pico Methyl-Seq Library Prep
Product: Zymo Pico Methyl Seq Library Prep
I am basing my protocol text off of Jill’s protocol, and you can also see Zymo’s protocol here and the general Putnam Lab protocol here.
Here’s the Pico Methyl-Seq library prep workflow:

Samples: All 10 extracted on 9/13
Samples were diluted to 10 ng in 20 uL of DNA elution buffer based on the gDNA tapestation concentrations.

Tapestation concentrations:
| sample_id | concentration | DIN |
|---|---|---|
| #4 (Frag A) | 4.86 ng/uL | 2.8 |
| #5 (Frag A) | 5.01 ng/uL | 2.8 |
| #8 (Frag B) | 4.46 ng/uL | 3.3 |
| #9 (Frag B) | 4.37 ng/uL | 3.1 |
| #15 (Frag C) | 3.80 ng/uL | 2.4 |
| #16 (Frag C) | 3.43 ng/uL | 2.5 |
| #20 (Frag D) | 3.37 ng/uL | 2.3 |
| #21 (Frag D) | 4.41 ng/uL | 2.8 |
| #26 (Frag E) | 2.75 ng/uL | - |
| #27 (Frag E) | 1.96 ng/uL | - |
| ng input | Starting volume (uL) | Volume DNA (uL) | Volume Tris (uL) |
|---|---|---|---|
| 10 | 20 | 2.1 | 17.9 |
| 10 | 20 | 2.0 | 18.0 |
| 10 | 20 | 2.2 | 17.8 |
| 10 | 20 | 2.3 | 17.7 |
| 10 | 20 | 2.6 | 17.4 |
| 10 | 20 | 2.9 | 17.1 |
| 10 | 20 | 3.0 | 17.0 |
| 10 | 20 | 2.3 | 17.7 |
| 10 | 20 | 3.6 | 16.4 |
| 10 | 20 | 5.1 | 14.9 |
Materials
- Kit contents
- PCR tubes, 1.5 mL tubes
- Thermocycler
- Heating block
- Mini centrifuge
- Vortex, shaker
- Aluminum beads (to keep things on ice)
- Magnetic stand for PCR tubes
- Custom index primers for Illumina if you have more than 6 samples (Zymo kit provides 6)
About indeces:
“1. Ensure that there is diversity within the first couple of bases. Illumina recommends choosing barcodes that do not share the same base at the position. 2. Order new primers according to the “Can additional index primers be purchased for multiplexing?” section 3. Illumina recommends the following multiplexing strategy for 6 or fewer samples: Pool of 2 samples: • Index #6 GCCAAT • Index #12 CTTGTA Pool of 3 samples: • Index #4 TGACCA • Index #6 GCCAAT • Index #12 CTTGTA Pool of 6 samples: • Index #2 CGATGT • Index #4 TGACCA • Index #5 ACAGTG • Index #6 GCCAAT • Index #7 CAGATC • Index #12 CTTGTA”
Kit contents
| Pico Methyl-Seq™ Library Prep Kit | D5456 (25 preps) | Storage Temperature |
|---|---|---|
| Lightning Conversion Reagent | 3 tubes | Room Temp. |
| M-Binding Buffer | 20 ml | Room Temp. |
| M-Wash Buffer* | 6 ml (conc.) | Room Temp. |
| L-Desulphonation Buffer | 10 ml | Room Temp. |
| DNA Elution Buffer | 2 ml | Room Temp. |
| PrepAmp Polymerase (13 U/μL) | 15 ul | -20 °C |
| PrepAmp Buffer (5X) | 75 µl | -20 °C |
| PrepAmp Primer (40 μM) | 30 µl | -20 °C |
| PrepAmp Pre-Mix | 120 µl | -20 °C |
| DNase/RNase-Free Water | 1 ml | Room Temp. |
| Zymo-Spin™ IC Columns | 100 | Room Temp. |
| Collection Tubes | 100 | Room Temp. |
| DNA Binding Buffer | 25 ml | Room Temp. |
| DNA Wash Buffer | 6 ml (conc.) | Room Temp. |
| LibraryAmp Master Mix (2X) | 625 µl | -20 °C |
| LibraryAmp Primers (10 μM) | 30 µl | -20 °C |
| Index Primer Sets - 6 Sets(10 μM) | 30 µl | -20 °C |
Protocol
Buffer preperation
Once buffers are prepared for a kit, they do not need to be prepared again.
- Add 26 mL of 95% EtOH to the M-Wash Buffer concentrate
- Add 26 mL of 95% EtOH to the DNA Wash Buffer concentrate
Best practices
- Preset the thermocycler programs
- Thaw and keep -80°C and -20°C components on ice unless instructed otherwise. Flick to mix and centrifuge before use
- Avoid multiple freeze-thaws, make aliquots if needed
- Calculate master mix volumes before library prepping (see spreadsheet here)
- Allow Kapa beads to equilibrate to room temperature >30 mins before use
- Resuspend magnetic particles immediately before each use by gently inverting until homogenous
Section 1: Bisulfite Conversion of DNA
- Thaw samples on ice
- Prepare samples with Tris buffer to 10ng in 20 uL in strip tubes
- Add 130 uL of Lightning Conversion Reagent to each tube
- Vortex for 10 seconds and centrifuge briefly
- Run thermocycler program (labeled BS CONVERSION PICO under Maggie’s profile)
- Store at 4°C for <20 hours (overnight)
Next Day:
- Prepare buffers if needed (see above)
- Label Zymo-spin IC columns (in collection tubes) and 1.5 mL tubes for each sample
- Section 1: 1 set of columns + 1 set of 1.5 mL tubes
- Section 2: 1 set of PCR tubes
- Section 3: 1 set of columns + 1 set of 1.5 mL tubes
- Section 4: 1 set of PCR tubes
- Section 4.5: 1 set of columns + 1 set of 1.5 mL tubes
- Section 5: 1 set of PCR tubes
- Bead Cleanup: 1 set of PCR tubes, labelled as final library (date, initals, info, etc)
- Total:
- 3 sets of columns + 1 set of 1.5 mL tubes
- 4 sets of PCR tubes, one labelled as final library
Section 1, continued
- Add 600 uL of M-Binding Buffer to each column
- Add the entire bisulfite-converted sample to column and invert several times to mix
- Centrifuge at 16,000 g for 30 seconds + discard flow through
- Add 100 uL M-Wash Buffer
- Centrifuge at 16,000 g for 30 seconds + discard flow through
- Add 200 uL L-Desulphonation Buffer to column and incubate at room temperature for 15 minutes
- Near end of incubation, heat DNA Elution buffer to 56°C on thermoblock
- After 15 min, centrifuge at 16,000 g for 30 seconds + discard flow through
- Add 200 uL of M-Wash Buffer
- Centrifuge at 16,000 g for 30 seconds + discard flow through
- Repeat the wash step above
- Move spin columns into their respective 1.5 mL tubes
- Add 8 uL of warmed DNA Elution buffer to column
- Incubate at room temperature for 3 minutes
- Centrifuge at 16,000 g for 30 seconds to elute bisulfite-converted DNA
- Check if all tubes have same volume (greater elution volume may cause library prep failure)
- Toss spin columns and keep 1.5 mL tubes on ice
Section 2: Amplification with PrepAmp Primer
Prepare Master mixes:
| Priming Master Mix (PMM) | PrepAmp Master Mix (PAMM) | “Diluted” PrepAmp Polymerase mix (dPAP) | 1st Amplification Master Mix (AMM) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Sample # (+5% error) | 5X PrepAmp Buffer | 40 uM PrepAmp Primer | 5X PrepAmp Buffer | PrepAmp Pre Mix | PrepAmp Polymerase | PrepAmp Polymerase | DNA Elution Buffer | Library Amp Mix 2X | Library Amp Primers 10 uM |
| 10.5 | 21 | 10.5 | 10.5 | 39.375 | 3.15 | 3.15 | 2.1 | 131.25 | 10.5 |
- Thaw the following reagents on ice
- PreAmpBuffer (5x)
- PreAmp Primer (40 uM)
- PreAmp Pre-mix
- PreAmp Polymerase
- Determine n number: number of samples + % error (~5%)
- Labeling scheme for master mixes
- PMM = A
- PAMM = B
- dPAP = C
- Create Priming Master Mix (PMM or A) on ice. Vortex and centrifuge briefly
- 2 uL 5x PreAmp Buffer * n =
- 1 uL 40 uM PreAmp Primer * n =
- Create PreAmp Master Mix (PAMM or B) on ice. Vortex and centrifuge briefly
- 1 uL 5x PrepAmp Buffer * n =
- 3.75 uL PrepAmp Pre Mix * n =
- 0.3 uL PrepAmp Polymerase * n =
- Create ‘diluted’ PreAmp Polymerase mix (dPAP or C) on ice. Pipette to mix and centrifuge briefly
- This is to avoid adding less than 0.5 uL during protocol. (original protocol asks you to add 0.3ul to the tubes in the thermocycler, sometimes that small of an amount does not come out of the tip so you can add DNA elution buffer to the enzyme to pipette 0.5ul).
- 0.3 uL PrepAmp Polymerase * n =
- 0.2 uL DNA Elution Buffer * n =
Amplification:
- On ice, add 7 uL of bisulfite converted sample to new PCR tubes
- Add 3 uL of PMM (A) to each tube
- Vortex and centrifuge briefly
Thermocycler settings:
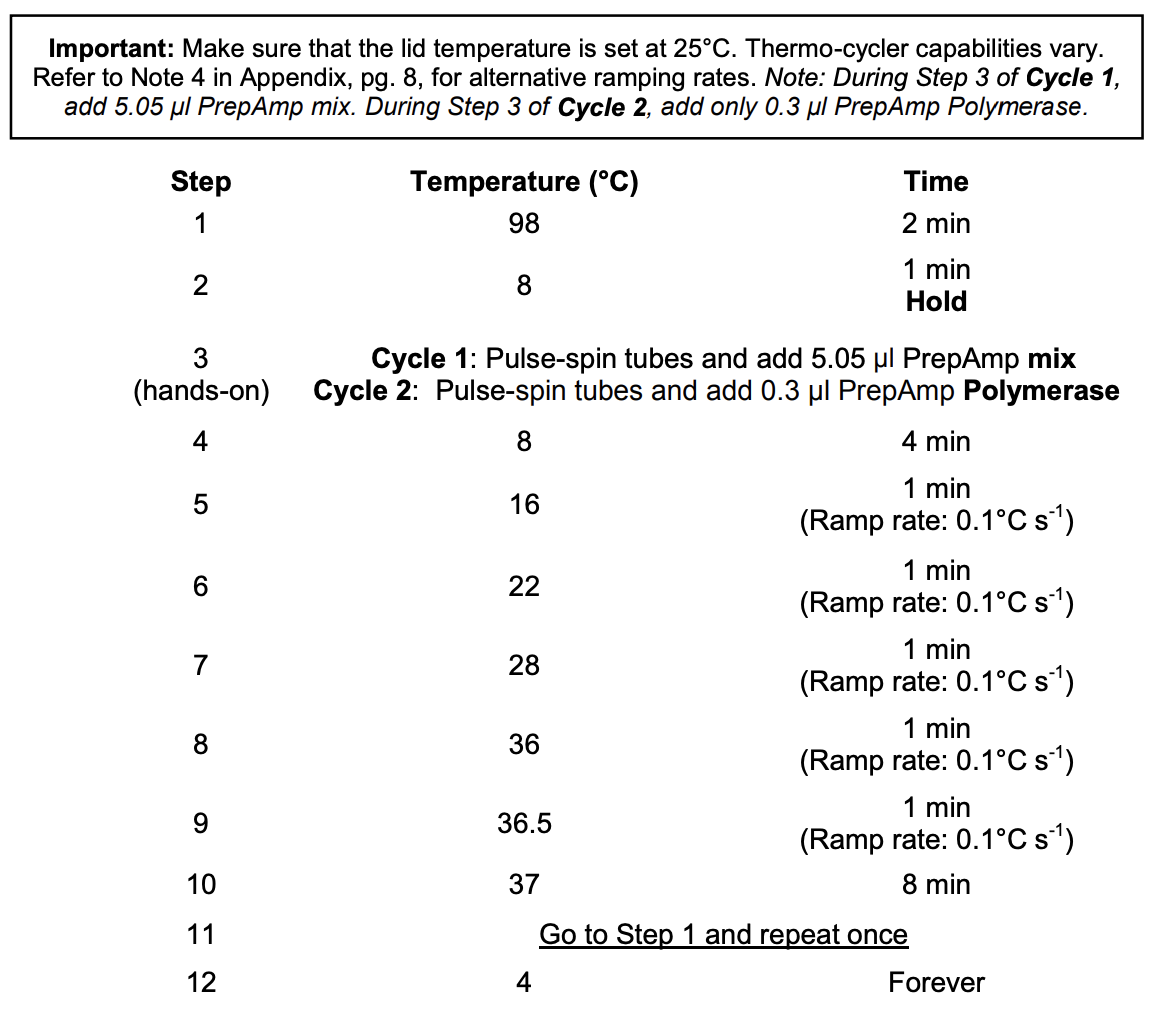
Lid should be set at 25°C
This program runs through two cycles. In the second step of each cycle, you need to add something to the tubes.
- Run the thermocycler program with the lid temperature at 25°C with 2 cycles
- During the Step 2 8°C hold:
- Cycle 1: Add 5.05 ul of PreAmp Master Mix (PAMM or B). Pipette up and down to mix, centrifuge briefly, put back in thermocycler and proceed past hold
- about 24 minutes later Cycle 2: Add 0.5 uL of diluted PreAmp Polymerase (dPAP or C) to each tube.Pipette up and down to mix, centrifuge briefly, put back in thermocycler and proceed past hold
- During the Step 2 8°C hold:
- Put samples on ice
- Current volume: 15.55 uL
Section 3: Purification with DNA Clean-up and Concentrator (DCC)
- Heat DNA Elution Buffer to 56°C on thermoblock
- Add 108.85 uL of DNA binding buffer (7:1 ratio of DNA binding buffer:sample) to PCR tube
- Vortex and spin down to mix, then transfer to spin column
- Centrifuge at 16,000 g for 30 seconds + discard flow through
- Add 200 uL of DNA Wash Buffer to each column
- Centrifuge at 16,000 g for 30 seconds + discard flow through
- Repeat this wash step
- Move spin columns into their respective 1.5 mL tubes
- Add 14 uL of warmed DNA Elution Buffer
- Incubate spin columns at room temperature for 2.5 minutes
- Centrifuge at 16,000 g for 30 seconds
- Toss spin columns and keep 1.5 mL tubes on ice
Section 4: Library Amplification
- Make Amplification Master Mix (AMM or D) on ice. Vortex and centrifuge briefly
- 12.5 uL 2X Library Amp Mix * n =
- 1 uL 10 uM Library Amp Primers * n =
- Add 13.5 of AMM to new strip tubes
- Add 11.5 uL of sample into each strip tube
- Vortex and centrifuge briefly
- Run the thermocycler program
Thermocycler settings:
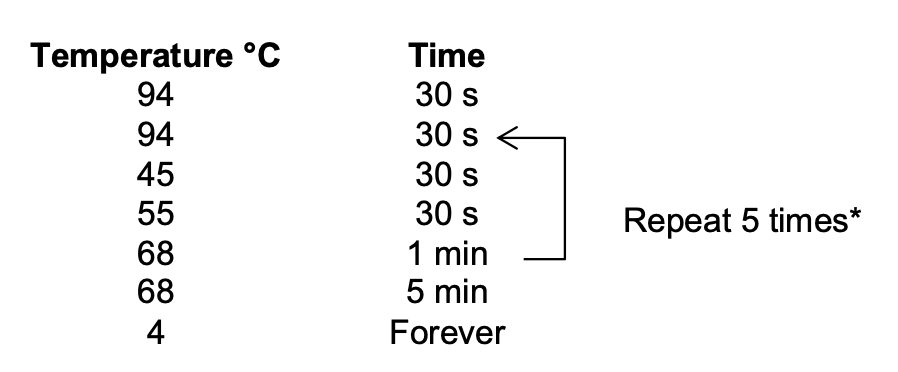
I will do 10 cycles. Possibly 12.
- Put samples on ice
- Current volume: 25 uL
Section 4.5: Purification with DNA Clean-up and Concentrator (DCC)
- Heat DNA Elution Buffer to 56°C on thermoblock
- Add 175 uL of DNA binding buffer (7:1 ratio of DNA binding buffer:sample)to PCR tube
- Vortex and spin down to mix, then transfer to spin column
- Centrifuge at 16,000 g for 30 seconds + discard flow through
- Add 200 uL of DNA Wash Buffer to each column
- Centrifuge at 16,000 g for 30 seconds + discard flow through
- Repeat this wash step
- Move spin columns to 1.5 mL tubes
- Add 14.5 uL of warmed DNA Elution Buffer to the column matrix
- Incubate for 2.5 minutes at room temperature
- Centrifuge at 16,000 g for 30 seconds
- Toss spin columns and keep 1.5 mL tubes on ice
Section 5: Amplification with Index Primers
Thermocycler settings:
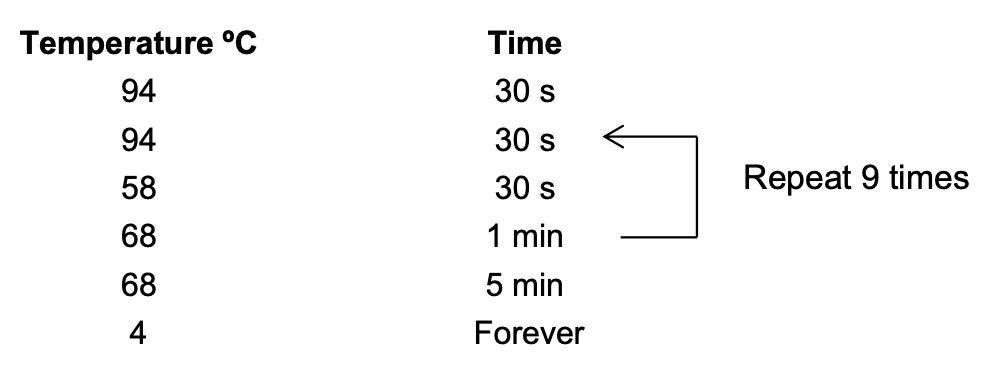
For our custom primers:
- Move 10.5 uL of sample into new strip tubes
- Add 12.5 uL of 2X Library Amp Master Mix to each sample
- Add 2 uL of combined i7 and i5 primer pair (1 uL of each)
- Run the thermocycler program for 10 amplification cycles
- Take Kapa beads out of fridge >30 minutes before next step
- After program is done, put samples on ice
For Zymo Index primers:
- Move 12 uL of sample into new strip tubes
- Add 12.5 uL of 2X Library Amp Master Mix to each sample
- Add 0.5 uL of index primer
- Run the thermocycler program for 10 amplification cycles
- Take Kapa beads out of fridge >30 minutes before next step
- After program is done, put samples on ice
1x Bead Cleanup
- Take Kapa beads out of fridge >30 minutes before using to allow beads to equilibrate to room temperature
- Shake the bottle to mix the beads (don’t vortex)
- Make fresh 80% ethanol using 100% ethanol
- Add 25 uL of beads to each sample
- Pipette up and down at least 10 times to mix sample and beads (or until homogeous)
- Put strip tubes on the shaker at 200 rpm for 15 minutes
- After 15 minutes, put tubes on magnetic stand
- Wait until liquid is clear (~3-5 minutes)
- Using a p200 set to 45 uL, remove the supernatent without disturbing the beads and discard
- Add 200 uL of 80% ethanol to each sample
- Remove ethanol from tube without disturbing beads and discard liquid
- Repeat the wash step
- Allow beads to air dry for 1-2 minutes
- Remove any residual liquid from each tube
- Resuspend beads in 15 uL of DNA Elution Buffer
- Put tubes back on shaker at 200 rpm for 5 minutes
- After 5 minutes, put tubes back on magnetic stand and wait until liquid is clear (~3-5 minutes)
- Move 15 uL of elute into new strip tubes
THIS IS THE FINAL WGBS LIBRARY. STORE AT -20°C.
QC
Run to visualize libraries. Here’s an example of what the library should look like on a Tapestation:
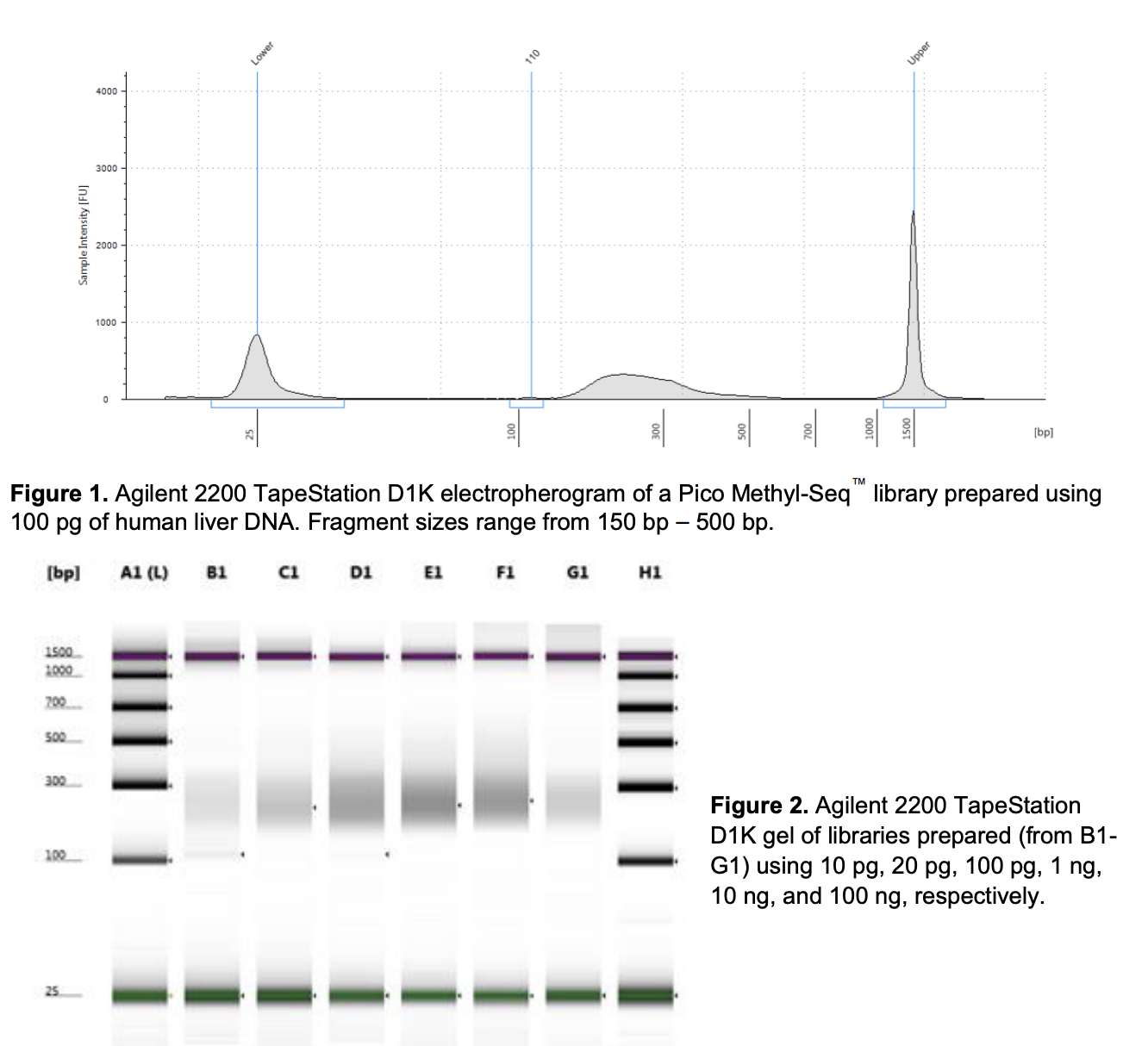
Library prep results, 9/17/24

These results look super odd! It could be primer dimer from having too little DNA input relative to primer and possibly too many cycles?
- While I did put in 10ng of DNA based on the tapestation values, keep in mind that the Qubit values (high sensitivity) were all non detectable, so it is unclear how much DNA really went into the prep.
“Primer dimers at 126 bp may appear as a result of a high PrepAmp Primer concentration relative to the amount of input DNA. The PrepAmp Primer (40 μM) can be diluted to 20 μM to alleviate this issue (Section 2).”
Full results can be found here
| Sample | Volume left after tapestation (uL) | Concentration (ng/uL) | ng Library (conc * 14 uL) |
|---|---|---|---|
| #4 (Frag A) | 14 | 4.01 | 56.14 |
| #5 (Frag A) | 14 | 1.6 | 22.4 |
| #8 (Frag B) | 14 | 2.41 | 33.74 |
| #9 (Frag B) | 14 | 4.84 | 67.76 |
| #15 (Frag C) | 14 | 0.523 | 7.322 |
| #16 (Frag C) | 14 | 3.38 | 47.32 |
| #20 (Frag D) | 14 | 2.06 | 28.84 |
| #21 (Frag D) | 14 | 0.931 | 13.034 |
| #26 (Frag E) | 14 | 7.56 | 105.84 |
| #27 (Frag E) | 14 | 1.69 | 23.66 |










Second bead cleanup of 9/17 libraries
I am attempting a 1.5X Kapa bead cleanup of the libraries above to try to remove the big peak at ~150 bp. Using the same protocol as the bead cleanup above.
- To each 14 uL library, thawed on ice, added 21 uL of beads and followed all the same steps.
- Tapestation basically didn’t change at all:
- concentration went down, peaks didn’t budge

| Sample | Volume left after tapestation (uL) | Concentration (ng/uL) | ng Library (conc * 14 uL) |
|---|---|---|---|
| #4 (Frag A) | 14 | 4.01 | 56.14 |
| #5 (Frag A) | 14 | 1.6 | 22.4 |
| #8 (Frag B) | 14 | 2.41 | 33.74 |
| #9 (Frag B) | 14 | 4.84 | 67.76 |
| #15 (Frag C) | 14 | 0.523 | 7.322 |
| #16 (Frag C) | 14 | 3.38 | 47.32 |
| #20 (Frag D) | 14 | 2.06 | 28.84 |
| #21 (Frag D) | 14 | 0.931 | 13.034 |
| #26 (Frag E) | 14 | 7.56 | 105.84 |
| #27 (Frag E) | 14 | 1.69 | 23.66 |
Full results can be found here, last two lanes are not related
Third bead cleanup of two 9/17 libraries
I am attempting a 0.9X Kapa bead cleanup on two of the libraries (#26 and 27) above to try to remove the big peak at ~150 bp. Using the same protocol as the bead cleanup above.
- To each 14 uL library, thawed on ice, increased volume to 30 uL with DNA elution buffer from kit, added 27 uL of beads and followed all the same steps.
- Tapestation basically didn’t change at all:
- just lost a ton of concentration, but 150 bp peak decreased a bit


| Sample | Volume left after tapestation (uL) | Concentration (ng/uL) | ng Library (conc * 14 uL) |
|---|---|---|---|
| #26 (Frag E) | 14 | 0.619 | 8.666 |
| #27 (Frag E) | 14 | 0.268 | 3.752 |
Full results can be found here, only the last two lanes
What’s next?
Start over with less cycles and less primer? 0.9X cleanup?
-
The peak is very likely strong adapter dimer.
-
but also, the concentrations are super low. so, need to figure out if I can increase cycles without just re-doing the adapter dimer /primer dimer problem.
- keep cycles same or even higher, but dilute LibraryAmp primers by 0.5x
Q8: Why does the library banding pattern look like a DNA ladder?
Using a higher ratio of PrepAmp Primer to DNA input can result in primer dimers that get carried through to the final library. To help avoid banding pattern: - Try reducing the amount of PrepAmp Primer to 20 nM (for inputs of ~10-50 ng) or 10 nM (for inputs <10 ng). - Please evaluate the best ratio for your sample type and sample amount. Ratio dependent on type of input (gDNA, restriction-enzyme digested, cfDNA, etc.). Are libraries produced by the Pico Methyl-Seq library prep kit directional or non-directional? Libraries prepared with this kit are non-directional and as such, the original-top, original-bottom, and the complementary strands for each will be represented.
- could also re-amp the already made library
- pippin prep for size selection
9/22/24: Try 2 of Library Prep, reducing adapter dimer and increasing amplification
- Testing the following modifications to the protocol:
- Dilute LibraryAmp primers by 0.5x
- Increased cycles in library amp from 10 to 17
Adapter dimer less than before but still very prominent peaks at 150 and 275 bp
- possible PCR bubble? 17 cycles maybe too many – but should I decrease library amp primer even further?
- if you look at the y axis of the tapestation graphs, the peaks are very similar to 9/17 prep but less high



| Sample | Volume left after tapestation (uL) | Concentration (ng/uL) | ng Library (conc * 14 uL) |
|---|---|---|---|
| #5 (Frag A) | 14 | 1.35 | 18.9 |
| #21 (Frag D) | 14 | 1.29 | 18.06 |
Full results can be found here
10/14/24: Try 3 of Library Prep with test sample (9/8/24 extraction), reducing adapter dimer and increasing amplification
- I chatted with Zymo and they recommended to dilute the PreAmp primer by 0.5X instead of the library amp primer and they also said not to use more than 12 cycles max. They also said to try to increase input as much as possible.
So, Jill set up sample #13 from 9/8/24 (thank you Jill!!) with 5 uL of DNA input. Based on the Tapestation concentration, this is an input of roughly 35 ng of DNA (3.5X what I used above), but Zymo also doesn’t trust the tapestation concentrations so who knows how much really went in.
Plan for day 2 library prep:
- Dilute PreAmp primer by 0.5X (0.5 uL H2O, 0.5 uL PreAmp Primer)
- Use 10 cycles in library amp step
Jill also made the following adjustments:
- Section 1 - incubation with warmed DNA elubtion buffer shortened from 5 to 3 minutes. Emma reported that she typically got less out of the column if she did the full 5 minutes.
- Section 3 - Incubation with warmed DNA elubtion buffer shortened from 5 to 2.5 minutes.
- Section 5 - incubation with warmed DNA elubtion buffer shortened from 5 to 2.5 minutes.
Results ! Much better!!
(THANK YOU JILL)
Adapter dimer less than before but still a tiny peak at ~250 bp


| Sample | Volume left after tapestation (uL) | Concentration (ng/uL) | ng Library (conc * 14 uL) |
|---|---|---|---|
| #13 (9/8/24) | 14 | 2.73 | 38.22 |
Redo plans
I want to redo all of my samples with these adjustments and increased DNA input. However, we need more D5000 reagents to QC the libraries so I am going to wait to start until next week.
11/19/24: Try 4 of Library Prep with Protocol Jill used on 10/15
| Sample | Volume left after tapestation (uL) | Concentration (ng/uL) | ng Library (conc * 14 uL) |
|---|---|---|---|
| #4 (9/13/24) | 14 | 0.857 | 11.998 |
| #8 (9/13/24) | 14 | 1.27 | 17.78 |
Did not work! Boo. Maybe my 9/13 samples are just too degraded compared to the one Jill used on 10/15 (from 9/8 extraction)


