
LCM Low Input RNA Library Prep
LCM Low Input RNA Library Prep
Using product: NEBNext® Single Cell/Low Input RNA Library Prep Kit for Illumina
Protocol: Link here. Most of the text here is taken directly from this protocol. I will only be using (and detailing the steps below) for one of the workflows in this protocol: “Section 2: Protocol for Low Input RNA: cDNA Synthesis, Amplification and Library Generation”. I successfully tested this on 7/30/24.
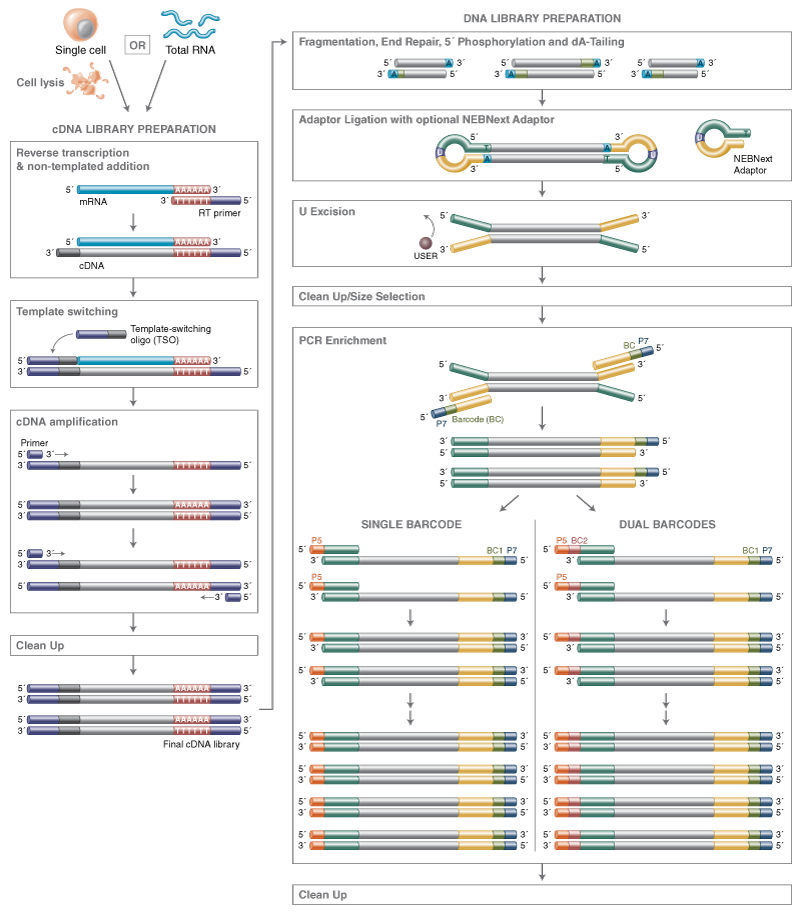
Overview image of workflow:
Materials
- NEBNext® Single Cell/Low Input RNA Library Prep Kit for Illumina
- Store entire kit at -20 ºC
- except NEBNext Cell Lysis Buffer, which, once prepared, should be stored at 4 ºC
- Store entire kit at -20 ºC
- DNase RNase free PCR strip tubes (USA Scientific 1402-1708)
- 80% Ethanol (freshly prepared)
- Nuclease-free water
- DNA LoBind Tubes (Eppendorf® #022431021)
- Magnetic rack or plate (e.g., NEBNext® Magnetic Separation Rack or equivalent)
- Thermocycler
- Vortex
- Microcentrifuge
- SPRIselect Reagent Kit (Beckman Coulter®, Inc. #B23317) or AMPure® XP Beads (Beckman Coulter, Inc. #A63881)
- we only have KAPA Pure beads
- Based on some research the recommended ratios seem really similar
- Agilent® Bioanalyzer® or similar fragment analyzer and associated consumables
- I will be using an Agilent Tapestation instead
- NEBNext Oligos
Kit contents
All reagents should be stored at –20°C. Colored bullets indicate the cap color of the reagent to be added to a reaction.
- ⚪️ Murine RNase Inhibitor (not used in the RNA-input kit)
- ⚪️ NEBNext Cell Lysis Buffer (once prepared, store at 4 ºC)
- 🟣 NEBNext Single Cell RT Primer Mix
- 🟣 NEBNext Single Cell RT Buffer
- 🟣 NEBNext Template Switching Oligo
- 🟣 NEBNext Single Cell RT Enzyme Mix
- 🟠 (or ⚪️) NEBNext Single Cell cDNA PCR Master Mix
- 🟠 NEBNext Single Cell cDNA PCR Primer
- 🟡 NEBNext Ultra II FS Enzyme Mix
- 🟡 NEBNext Ultra II FS Reaction Buffer
- 🔴 NEBNext Ultra II Ligation Master Mix
- 🔴 NEBNext Ligation Enhancer
- 🔵 NEBNext Ultra II Q5® Master Mix
- ⚪️ NEBNext Bead Reconstitution Buffer
- ⚪️ NEBNext Adaptor Dilution Buffer
- ⚪️ TE Buffer
- ⚪️ Nuclease-free Water
Samples: All 10 extracted on 9/13

Tapestation concentrations and DV200 values:
| sample_id | concentration | RIN | DV200 |
|---|---|---|---|
| #4 (Frag A) | 333 pg/uL (0.333 ng/uL) | - | 59.70% |
| #5 (Frag A) | 271 pg/uL (0.271 ng/uL) | - | 61.63% |
| #8 (Frag B) | 246 pg/uL (0.246 ng/uL) | - | 67.53% |
| #9 (Frag B) | 288 pg/uL (0.288 ng/uL) | - | 63.35% |
| #15 (Frag C) | 275 pg/uL (0.275 ng/uL) | - | 59.40% |
| #16 (Frag C) | 263 pg/uL (0.263 ng/uL) | - | 66.72% |
| #20 (Frag D) | 272 pg/uL (0.272 ng/uL) | - | 66.19% |
| #21 (Frag D) | 339 pg/uL (0.339 ng/uL) | - | 60.31% |
| #26 (Frag E) | 283 pg/uL (0.283 ng/uL) | - | 61.44% |
| #27 (Frag E) | 211 pg/uL (0.211 ng/uL) | - | 72.20% |
| Tapestation concentration | Library Prep volume input | Library Prep ng input |
|---|---|---|
| 0.333 | 8 | 2.66 |
| 0.271 | 8 | 2.17 |
| 0.246 | 8 | 1.97 |
| 0.288 | 8 | 2.30 |
| 0.275 | 8 | 2.20 |
| 0.263 | 8 | 2.10 |
| 0.272 | 8 | 2.18 |
| 0.339 | 8 | 2.71 |
| 0.283 | 8 | 2.26 |
| 0.211 | 8 | 1.69 |
Median input: 2.19 ng. An input of 8uL was used for all samples
Protocol for Low Input RNA: cDNA Synthesis, Amplification and Library Generation
Sample Recommendations
- The RNA sample should be free of salts (e.g., Mg2+, or guanidinium salts), divalent cation chelating agents (e.g. EDTA, EGTA, citrate), or organics (e.g., phenol and ethanol). If an excess amount of genomic DNA is present in RNA samples, an optional DNase I treatment could be performed. Inactivate/remove DNase I after treatment.
- Assess quality of the input RNA by running input RNA on an Agilent Bioanalyzer to determine the RNA Integrity Number (RIN).
- Starting Material: 2 pg–200 ng poly(A) tail-containing total RNA (DNA free), RIN score ≥ 8.0.
- Our RNA is definitely not RIN score > 8.0, but we will see what we can do.
2.1 Sample and Reagent preperation
- Day 1 (Annealing, RT, cDNA PCR, cleanup, cDNA QC):
- Prepare fresh 80% Ethanol with nucelase-free water
- Thaw total RNA on ice prior to starting the protocol
- Briefly centrifuge the tubes containing 🟣 NEBNext Single Cell RT Enzyme Mix to collect solutions to the bottom of the tubes, then place on ice.
- Thaw the following frozen components at room temperature
- ⚪️ NEBNext Cell Lysis Buffer (once prepared, store at 4 ºC)
- if the ⚪️ 10X NEBNext Cell Lysis Buffer appears cloudy after thawing, incubate briefly at 37°C to clear up the solution.
- 🟣 NEBNext Single Cell RT Primer Mix
- 🟣 NEBNext Single Cell RT Buffer
- 🟣 NEBNext Template Switching Oligo
- 🟠 (or ⚪️) NEBNext Single Cell cDNA PCR Master Mix
- 🟠 NEBNext Single Cell cDNA PCR Primer
- ⚪️ NEBNext Bead Reconstitution Buffer
- ⚪️ TE Buffer
- ⚪️ Nuclease-free Water
- ⚪️ NEBNext Cell Lysis Buffer (once prepared, store at 4 ºC)
- Mix each component thoroughly, centrifuge briefly to collect solutions to the bottom of the tube, and then place on ice.
- Leave the NEBNext Cell Lysis Buffer bottle at 4°C or at room temperature for storage.
- Day 2 (Fragmentation/End Prep, Adaptor Ligation, Cleanup of Adaptor-ligated DNA, PCR enrichment, cleanup, and final library QC):
- Prepare fresh 80% Ethanol with nucelase-free water
- Thaw the following frozen components at room temperature
- 🟡 NEBNext Ultra II FS Enzyme Mix
- 🟡 NEBNext Ultra II FS Reaction Buffer
- 🔴 NEBNext Ultra II Ligation Master Mix
- 🔴 NEBNext Ligation Enhancer
- 🔵 NEBNext Ultra II Q5® Master Mix
- ⚪️ NEBNext Adaptor Dilution Buffer
- ⚪️ TE Buffer
- ⚪️ Nuclease-free Water
- Mix each component thoroughly, centrifuge briefly to collect solutions to the bottom of the tube, and then place on ice.
Day 1 - 9/18/2024
2.2. Primer Annealing for First Strand Synthesis
Note, this is for each RNA sample individually in an individual PCR tube
-
2.2.1. To anneal cDNA Primer with total RNA samples, prepare the reaction as follows (on ice):
- for an input of 4.99 ng RNA, at 8µl volume, the sample would have a minimum concentration of 0.62 ng/µl
- Most of our high quality RNA is below this concentration, so we will use the < 5 ng mixture
- for a low-concentration sample (like LCM RNA), of lets say, 0.280 ng/µl, 8 µl of input would be 2.24 ng. So we would put in 8 µl of the thawed RNA and use the < 5 ng mixture.
- if you have > 7 µl of an RNA of concentration greater than 0.714 ng/µl (5 ng / 7 µl), you should use the ≥ 5 ng RNA workflow.
| COMPONENT | < 5 ng RNA: VOLUME (µl) | ≥ 5 ng RNA: VOLUME (µl) |
|---|---|---|
| Total RNA | Up to 8 µl | Up to 7 µl |
| 🟣 NEBNext Single Cell RT Primer | 1 µl | 2 µl |
| Nuclease-free Water | Variable | Variable |
| Total Volume | 9 µl | 9 µl |
- 2.2.2. Mix well by pipetting up and down gently at least 10 times, then centrifuge briefly to collect solution to the bottom of the tubes.
- 2.2.3. Incubate for 5 minutes at 70°C in a thermal cycler with the heated lid set to 105°C, then hold at 4ºC until next step.
During the above annealing step, prepare the components for the following step.
2.3. Reverse Transcription (RT) and Template Switching
- 2.3.1. Vortex the NEBNext Single Cell RT Buffer briefly
- Note: It is important to vortex the buffer prior to use for optimal performance (they even have a video about this)
- Then, prepare the RT mix in a separate tube as follows (adding NEBNext Single Cell RT Enzyme Mix last)
| COMPONENT | VOLUME (µl) PER REACTION |
|---|---|
| 🟣 NEBNext Single Cell RT Buffer (VORTEXED) | 5 µl |
| 🟣 NEBNext Template Switching Oligo | 1 µl |
| 🟣 NEBNext Single Cell RT Enzyme Mix | 2 µl |
| Nuclease-free Water | 3 µl |
| Total Volume | 11 µl |
- 2.3.2. Mix thoroughly by pipetting up and down several times, then centrifuge briefly to collect solutions to the bottom of tubes.
- 2.3.3. Combine 11 μl of the RT mix (above) with 9 μl of the annealed sample (Step 2.2.3.).
- Mix well by pipetting up and down at least 10 times and centrifuge briefly.
- 2.3.4. Incubate the reaction mix in a thermal cycler with the following steps and the heated lid set to 105°C:
- 90 minutes at 42°C
- 10 minutes at 70°C
- Hold at 4°C
🛑 Safe Stopping Point: Samples can be safely stored overnight at 4°C or –20°C.
2.4. cDNA Amplification by PCR
- 2.4.1. Prepare cDNA amplification mix as follows:
- ⚪️ NEBNext Cell Lysis Buffer (10X) needs to be homogenous with no precipitates (should be clear, if not, warm to 37 ºC). Vortex thoroughly and spin down before adding.
| COMPONENT | VOLUME (µl) PER REACTION |
|---|---|
| 🟠 (or ⚪️) NEBNext Single Cell cDNA PCR Master Mix | 50 µl |
| 🟠 NEBNext Single Cell cDNA PCR Primer | 2 µl |
| ⚪️ NEBNext Cell Lysis Buffer (10X) | 0.5 µl |
| Nuclease-free Water | 27.5 µl |
| Total Volume | 80 µl |
- 2.4.2. Add 80 µl cDNA amplification mix to 20 µl of the sample from Step 2.3.4.
- Mix by pipetting up and down at least 10 times.
- 2.4.3. Incubate the reaction in a thermal cycler with the following PCR cycling conditions and the heated lid set to 105°C:
| CYCLE STEP | TEMP | TIME | CYCLES |
|---|---|---|---|
| Initial Denaturation | 98°C | 45 seconds | 1 |
| Denaturation | 98°C | 10 seconds | 7–21 depending on RNA input (see manual) |
| Annealing | 62°C | 15 seconds | ^ |
| Extension | 72°C | 3 minutes | ^ |
| Final Extension | 72°C | 5 minutes | 1 |
| Hold | 4°C | ∞ |
This step should take about an hour depending on # of cycles.
For 1-10 ng of RNA, they recommend 10-11 cycles.
Based on looking at other Putnam lab Library Prep protocols, we almost always have to increase the number of cycles for our input. And mine is extra degraded. I will increase cycles to 15.
For the various inputs listed above, the recommended PCR cycles will typically result in cDNA yields between 1–20 ng (in most cases 5–15 ng). We recommend quantifying cDNA after the cleanup (Step 2.5) before proceeding to the library preparation (Sections 2.7–2.12).
🛑 Safe Stopping Point: Samples can be safely stored overnight at 4°C or –20°C.
2.5. Cleanup of Amplified cDNA
This should be done at post-PCR bench
- 2.5.1. Allow the ⚪️ NEBNext Bead Reconstitution Buffer and the SPRI® beads (if stored at 4°C) to warm to room temperature for at least 30 minutes before use.
- Shake SPRI Beads to resuspend well
- Prepare fresh 80% ethanol
- 800 uL per sample
- Prepare 0.1X TE (dilute ⚪️ 1X TE Buffer 1:10 in nuclease free water)
- 50 uL per sample
- 2.5.2. Add 60 μl (0.6X of sample volume) resuspended beads to the PCR reaction.
- Mix well by pipetting up and down at least 10 times.
- Be careful to expel all of the liquid out of the tip during the last mix
- 2.5.3. Incubate samples on the bench top for at least 5 minutes at room temperature.
- 2.5.4. Place the tube/plate on an appropriate magnetic stand to separate the beads from the supernatant. If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing on the magnetic stand.
- 2.5.5. After 5 minutes (or when the solution is clear), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain cDNA (Caution: do not discard the beads).
- 2.5.6. Add 200 μl of 80% freshly prepared ethanol to the tube/plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant. Be careful not to disturb the beads that contain cDNA.
- 2.5.7. Repeat Step 2.5.6. once for a total of two washes. Be sure to remove all visible liquid after the second wash. If necessary, briefly spin the tube/plate, place back on the magnet and remove traces of ethanol with a p10 pipette tip.
- 2.5.8. Air dry the beads for up to 5 minutes while the tube/plate is on the magnetic stand with the lid open.
- Caution: Do not over-dry the beads. This may result in lower recovery of cDNA. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack, they are too dry.
- 2.5.9. Remove the tube/plate from the magnetic stand.
- Elute the cDNA from the beads by adding 50 μl of 0.1X TE (dilute ⚪️ 1X TE Buffer 1:10 in nuclease free water).
- 2.5.10. Mix well by pipetting up and down 10 times, or on a vortex mixer. Incubate for at least 2 minutes at room temperature.
- If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells.
- 2.5.11. Add 45 μl of (room temperature) ⚪️ NEBNext Bead Reconstitution Buffer to the eluted cDNA + bead mixture from Step 2.5.10 for a second sample clean up.
- Mix well by pipetting up and down at least 10 times
- 2.5.12. Incubate samples on the bench top for at least 5 minutes at room temperature.
- 2.5.13. Place the tube/plate on an appropriate magnetic stand to separate the beads from the supernatant. If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing on the magnetic stand.
- 2.5.14. After 5 minutes (or when the solution is clear), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain cDNA (Caution: do not discard the beads).
- 2.5.15. Add 200 μl of 80% freshly prepared ethanol to the tube/plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant. Be careful not to disturb the beads that contain cDNA.
- 2.5.16. Repeat Step 2.5.15 once for a total of two washes. Be sure to remove all visible liquid after the second wash. If necessary, briefly spin the tube/plate, place back on the magnet and remove traces of ethanol.
- 2.5.17. Air dry the beads for up to 5 minutes while the tube/plate is on the magnetic stand with the lid open.
- Caution: Do not over-dry the beads. This may result in lower recovery of cDNA. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack, they are too dry.
- 2.5.18. Remove the tube/plate from the magnetic stand.
- Elute the cDNA from the beads by adding 33 μl of ⚪️ 1X TE (provided in kit).
- 2.5.19. Mix well by pipetting up and down 10 times, or on a vortex mixer. Incubate for at least 2 minutes at room temperature.
- If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing back on the magnetic stand.
NOW WE KEEP SUPERNATANT
- 2.5.20. Place the tube/plate on the magnetic stand. After 5 minutes (or when the solution is clear), transfer 30 μl to a new PCR tube.
2.6. Assess Amplified cDNA Quality and Quantity on a Bioanalyzer
- 2.6.1. Run 1 µl of amplified cDNA from Step 2.5.20 on a DNA High Sensitivity Chip.
- I will use DNA tapestation instead
- 1 ng–20 ng cDNA yield is typical yield, with broad peak around 2000 bp
- See manual for recommendations based on QC results.
- While 1 ng–20 ng cDNA yield is typical, 100 pg–20 ng purified cDNA can be used in the library construction protocol (Sections 2.7–2.12). If using cDNA outside the range of 1 ng–20 ng (as determined in Step 2.6), adjust the PCR cycles to amplify the adaptor ligated DNA. For details, see Step 2.10 in this protocol.
- If the cDNA yield is variable, the samples can be normalized to the same concentration prior to Step 2.7 in order to treat all of the samples with the same number of PCR cycles.
🛑 Safe Stopping Point: Samples can be safely stored overnight at 4°C or –20°C.
Day 2 - 9/19/2024
2.7. Fragmentation/End Prep
- 2.7.1. Ensure that the 🟡 NEBNext Ultra II FS Reaction Buffer is completely thawed. If a precipitate is seen in the buffer, pipette up and down several times to break it up, and quickly vortex to mix. Place on ice until use.
- 2.7.2. Vortex the 🟡 NEBNext Ultra II FS Enzyme Mix 5–8 seconds prior to use and place on ice.
- 2.7.3. Add the following components to a 0.2 ml thin wall PCR tube on ice:
| COMPONENT | VOLUME (µl) PER REACTION |
|---|---|
| cDNA (Step 2.5.20) | 26 µl |
| 🟡 NEBNext Ultra II FS Reaction Buffer | 7 µl |
| 🟡 NEBNext Ultra II FS Enzyme Mix | 2 µl |
| Total Volume | 35 µl |
- 2.7.4. Vortex the reaction for 5 seconds and briefly spin in a microcentrifuge.
- 2.7.5. In a thermal cycler, with the heated lid set to 75°C, run the following program:
- 25 minutes at 37°C
- 30 minutes at 65°C
- Hold at 4°C
If necessary, can stop here and put at -20ºC, but you should keep going with Adaptor Ligation if possible
2.8. Adaptor Ligation
- From the NEB Oligo Kit, thaw adaptor for illumina NEBNext Adaptor for Illumina and USER® Enzyme
- 2.8.1. Dilute NEBNext Adaptor for Illumina by 25-fold (0.6 µM) in the NEBNext ⚪️ Adaptor Dilution Buffer.
- 2.8.2. Mix the 🔴 NEBNext Ultra II Ligation Master Mix by pipetting up and down several times.
- 2.8.3. Add the following components directly to the FS Reaction Mixture on ice:
Note: Do not mix these components ahead of time. Add straight to strip tube (that just came out of thermocycler) on ice. Caution: NEBNext Ultra II Ligation Master Mix is very viscous, pipette with care.
| COMPONENT | VOLUME (µl) PER REACTION |
|---|---|
| FS Reaction Mixture (Step 2.7.5) | 35 µl |
| 🔴 NEBNext Ultra II Ligation Master Mix | 30 µl |
| 🔴 NEBNext Ligation Enhancer | 1 µl |
| 🔴 NEBNext Adaptor for Illumina (diluted 1:25) | 2.5 µl |
| Total Volume | 68.5 µl |
- 2.8.4. Set a 100 μl or 200 μl pipette to 50 μl and then pipette the entire volume up and down at least 10 times to mix thoroughly.
- Perform a quick spin to collect all liquid from the sides of the tube. (Caution: The NEBNext Ultra II Ligation Master Mix is very viscous. Care should be taken to ensure adequate mixing of the ligation reaction, as incomplete mixing will result in reduced ligation efficiency. The presence of a small amount of bubbles will not interfere with performance).
- 2.8.5. Incubate at 20°C for 15 minutes in a thermal cycler with the heated lid off.
- 2.8.6. Add 3 μl of 🔴 USER® Enzyme to the ligation mixture from Step 2.8.5.
- 2.8.7. Mix well and incubate at 37°C for 15 minutes with the heated lid set to ≥ 47°C.
🛑 Safe Stopping Point: Samples can be safely stored overnight at 4°C or –20°C.
2.9. Cleanup of Adaptor-ligated DNA
- 2.9.1. If stored at 4°C allow the SPRI beads to warm to room temperature for at least 30 minutes before use.
- Shake SPRI beads to resuspend well
- Prepare fresh 80% ethanol
- 400 uL per sample
- Prepare 0.1X TE (dilute ⚪️ 1X TE Buffer 1:10 in nuclease free water)
- 17 uL per sample
- 2.9.2. Add 57 μl (0.8X of sample volume) resuspended beads to the PCR reaction.
- Mix well by pipetting up and down at least 10 times.
- Be careful to expel all of the liquid out of the tip during the last mix.
- 2.9.3. Incubate samples on the bench top for at least 5 minutes at room temperature.
- 2.9.4. Place the tube/plate on an appropriate magnetic stand to separate the beads from the supernatant. If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing on the magnetic stand.
- 2.9.5. After 5 minutes (or when the solution is clear), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets (Caution: do not discard the beads).
- 2.9.6. Add 200 μl of 80% freshly prepared ethanol to the tube/ plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets.
- 2.9.7. Repeat Step 2.9.6 once for a total of two washes. Be sure to remove all visible liquid after the second wash. If necessary, briefly spin the tube/plate, place back on the magnet and remove traces of ethanol with a p10 pipette tip.
- 2.9.8. Air dry the beads for up to 5 minutes while the tube/plate is on the magnetic stand with the lid open.
- Caution: Do not over-dry the beads. This may result in lower recovery of DNA. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack, they are too dry.
- 2.9.9. Remove the tube/plate from the magnetic stand.
- Elute the DNA target from the beads by adding 17 μl of 0.1X TE (dilute ⚪️ 1X TE Buffer 1:10 in nuclease free water).
- 2.9.10. Mix well by pipetting up and down 10 times, or on a vortex mixer. Incubate for at least 2 minutes at room temperature.
- If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing back on the magnetic stand.
NOW WE KEEP SUPERNATANT
- 2.9.11. Place the tube/plate on the magnetic stand. After 5 minutes (or when the solution is clear), transfer 15 μl to a new PCR tube. Proceed to PCR Enrichment of Adaptor-ligated DNA in Section 2.10.
🛑 Safe Stopping Point: Samples can be safely stored overnight at 4°C or –20°C.
2.10. PCR Enrichment of Adaptor-ligated DNA
- From the NEB Oligo Kit, thaw i7 and i5 primers according to instructions
- 2.10.1. Combine the following components in a sterile tube:
- Using Option A, since my NEBNext oligo kit has index primers supplied in tubes. These kits have the forward and reverse primers supplied in separate tubes. Primers are supplied at 10 µM each.
- *Refer to the corresponding NEBNext Oligo kit manual for determining valid barcode combinations.
- **Use only one i7 primer/ index primer per sample. Use only one i5 primer (or the universal primer for single index kits) per sample
| COMPONENT | VOLUME (µl) PER REACTION |
|---|---|
| Adaptor Ligated DNA Fragments (Step 2.9.11.) | 15 µl |
| 🔵 NEBNext Ultra II Q5® Master Mix | 25 µl |
| 🔵 Index Primer/i7 Primer*,** | 5 µl |
| 🔵 Universal PCR Primer/i5 Primer*, ** | 5 µl |
| Total Volume | 50 µl |
- 2.10.2. Set a 100 µl or 200 μl pipette to 40 μl and then pipette the entire volume up and down at least 10 times to mix thoroughly.
- Perform a quick spin to collect all liquid from the sides of the tube.
- 2.10.3. Place the tube on a thermal cycler and perform PCR amplification using the following PCR cycling conditions:
If your cDNA input is outside the input range of 1 ng–20 ng, adjust the PCR cycle numbers accordingly. We recommend a minimum of 3 PCR cycles for all of the original molecules to make it into the final library. For cDNA yield of 100 pg we recommend testing 12 PCR cycles. For cDNA input of 1 ng–20 ng, the typical Illumina library yield, using 8 PCR cycles, is 100 ng–1 μg.
| CYCLE STEP | TEMP | TIME | CYCLES |
|---|---|---|---|
| Initial Denaturation | 98°C | 30 seconds | 1 |
| Denaturation | 98°C | 10 seconds | 8 *dependent on cDNA input |
| Annealing/Extension | 65°C | 75 seconds | ^ |
| Final Extension | 65°C | 5 minutes | 1 |
| Hold | 4°C | ∞ |
2.11. Cleanup of PCR Reaction
- 2.11.1. If stored at 4°C allow the SPRI beads to warm to room temperature for at least 30 minutes before use.
- Shake SPRI beads to resuspend well
- Prepare fresh 80% ethanol
- 400 uL per sample
- Prepare 0.1X TE (dilute ⚪️ 1X TE Buffer 1:10 in nuclease free water)
- 33 uL per sample
- 2.11.2. Add 45 μl (0.9X of sample volume) resuspended beads to the PCR reaction.
- Mix well by pipetting up and down at least 10 times.
- Be careful to expel all of the liquid out of the tip during the last mix.
- 2.11.3. Incubate samples on bench top for at least 5 minutes at room temperature.
- 2.11.4. Place the tube/plate on an appropriate magnetic stand to separate the beads from the supernatant. If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing on the magnetic stand.
- 2.11.5. After 5 minutes (or when the solution is clear), carefully remove and discard the supernatant.
- Be careful not to disturb the beads that contain DNA targets (Caution: do not discard the beads).
- 2.11.6. Add 200 μl of 80% freshly prepared ethanol to the tube/ plate while in the magnetic stand. Incubate at room temperature for 30 seconds, and then carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets.
- 2.11.7. Repeat Step 2.11.6 once for a total of two washes. Be sure to remove all visible liquid after the second wash. If necessary, briefly spin the tube/plate, place back on the magnet and remove traces of ethanol with a p10 pipette tip.
- 2.11.8. Air dry the beads for up to 5 minutes while the tube/plate is on the magnetic stand with the lid open.
- Caution: Do not over-dry the beads. This may result in lower recovery of DNA. Elute the samples when the beads are still dark brown and glossy looking, but when all visible liquid has evaporated. When the beads turn lighter brown and start to crack, they are too dry.
- 2.11.9. Remove the tube/plate from the magnetic stand.
- Elute the DNA target from the beads by adding 33 μl of 0.1X TE (dilute ⚪️ 1X TE Buffer 1:10 in nuclease free water).
- 2.11.10. Mix well by pipetting up and down 10 times, or on a vortex mixer. Incubate for at least 2 minutes at room temperature.
- If necessary, quickly spin the sample to collect the liquid from the sides of the tube or plate wells before placing back on the magnetic stand.
NOW WE KEEP SUPERNATANT
- 2.11.11. Place the tube/plate on the magnetic stand. After 5 minutes (or when the solution is clear), transfer 30 μl to a new PCR tube.
Libraries can be stored at –20°C.
2.12. Assess Library Quality and Quantity on a Bioanalyzer
- 2.12.1. Dilute library (from Step 2.11.11) 5-fold in 0.1X TE Buffer (dilute ⚪️ 1X TE Buffer 1:10 in nuclease free water)
- Mix 1 uL library with 4 uL 0.1X TE
- inputs ≤ 1 ng may not require dilution to run on a Bioanalyzer.
- 2.12.2. Run 1 μl on a DNA High Sensitivity Chip.
- I am going to try with tapestation
- 2.12.3. Check that the electropherogram shows a narrow distribution with a peak size of 300–350 bp.
- Note: If a peak ~80 bp (primers) or 128-140 bp (adaptor-dimer) is visible in the Bioanalyzer trace, bring up the sample volume (from Step 2.11.11.) to 50 μl with 0.1X TE Buffer and repeat the cleanup of PCR Reaction as described in Section 2.11. You may see adaptor-dimer when starting with inputs ≤ 1 ng.
Typical Yield of Illumina Library from a Reaction
Actual yields will depend on the quality and quantity of the input cDNA. Typical library yields range between 100 ng–1 μg based on the PCR cycle recommendations provided in Section 2.10.
9/19/24 cDNA Results


cDNA and planned cycles/indeces
| Sample Type | cDNA concentration | amount cDNA input (conc * 26 uL) | PCR cycles for final step | Index_ID | index |
|---|---|---|---|---|---|
| #4 (Frag A) | 0.0869 | 2.2594 | 11 | E7500S-21 | GTTTCG |
| #5 (Frag A) | 0.211 | 5.486 | 9 | E7500S-14 | AGTTCC |
| #8 (Frag B) | 0.0744 | 1.9344 | 11 | E7500S-23 | GAGTGG |
| #9 (Frag B) | 0.0232 | 0.6032 | 11 | E7500S-18 | GTCCGC |
| #15 (Frag C) | 0.199 | 5.174 | 9 | E7500S-25 | ACTGAT |
| #16 (Frag C) | 0.172 | 4.472 | 9 | E7500S-20 | GTGGCC |
| #20 (Frag D) | 0.24 | 6.24 | 9 | E7500S-13 | AGTCAA |
| #21 (Frag D) | 0.112 | 2.912 | 9 | E7500S-15 | ATGTCA |
| #26 (Frag E) | 0.223 | 5.798 | 9 | E7500S-22 | CGTACG |
| #27 (Frag E) | 0.188 | 4.888 | 9 | E7500S-19 | GTGAAA |
#5s 5 and 15 were tested on 9/18/24 (last two lanes) with 17 cycles to make sure I could get adequate amplification. I cleaned and QC’d these on 9/18/24 and then set up the other 8 reactions on 9/18/24 as well, also with 17 cycles for cDNA amplification. The other 8 were cleaned and QC’d on 9/19/24. After all cDNA was QC’d, I determined how many cycles would be used for the final library amplification step, and decided to use 11 cycles for cDNA input < 3 ng (#s 4, 8, and 9) and 9 cycles for all the rest.
Full results can be found here.
9/20/24 Final Library Results
These look absolutely amazing. In the future any cDNA concentration of less than 5 ng/uL could probably benefit from 11 insterad of 9 cycles but these look so so good! yay!


Full results can be found here and here (not scaled by sample). Last two lanes are from a different project.
| Sample Type | Final library concentrations, Tapestation | ng Library (conc * 29 uL) |
|---|---|---|
| #4 (Frag A) | 24.3 | 704.7 |
| #5 (Frag A) | 32.5 | 942.5 |
| #8 (Frag B) | 26.5 | 768.5 |
| #9 (Frag B) | 12 | 348 |
| #15 (Frag C) | 11.9 | 345.1 |
| #16 (Frag C) | 5.14 | 149.06 |
| #20 (Frag D) | 10.3 | 298.7 |
| #21 (Frag D) | 3.52 | 102.08 |
| #26 (Frag E) | 9.3 | 269.7 |
| #27 (Frag E) | 6.42 | 186.18 |
Notes
Thermocycler programs to program:
- Primer Annealing (2.2.3):
- 5 minutes at 70°C (with heated lid set to 105°C)
- hold at 4ºC until next step
- RT (2.3.4):
- 90 minutes at 42°C (with heated lid set to 105°C)
- 10 minutes at 70°C (with heated lid set to 105°C)
- Hold at 4°C
- cDNA Amplification (2.4.3):
- 98°C, 45 seconds (with heated lid set to 105°C)
- 7-21 cycles (likely 15-17) of
- 98°C, 10 seconds (with heated lid set to 105°C)
- 62°C, 15 seconds (with heated lid set to 105°C)
- 72°C, 3 minutes (with heated lid set to 105°C)
- 72°C, 5 minutes (with heated lid set to 105°C)
- Hold at 4°C
- Fragmentation (2.7.5):
- 25 minutes at 37°C (with heated lid set to 75°C)
- 30 minutes at 65°C (with heated lid set to 75°C)
- Hold at 4°C
- Adaptor Ligation 1 (2.8.5):
- 15 minutes at 20°C (no heated lid - set OFF)
- Adaptor Ligation 2 (2.8.7):
- 15 minutes at 37°C (with heated lid set to ≥ 47°C)
- PCR Enrichment (2.10.3):
- no info about heated lid
- 98°C, 30 seconds
- 8 cycles (dept. on cDNA) of
- 98°C, 10 seconds
- 65°C, 75 seconds
- 65°C, 5 minutes
- Hold at 4°C
Research about adaptors and if we can use custom ones or not – we ended up purchasing one of the NEB Single Index Oligo sets
NEBNext Adaptor for Illumina Overview
NEBNext Adaptor for Illumina sequence:
5´-/5Phos/GAT CGG AAG AGC ACA CGT CTG AAC TCC AGT CdUA CAC TCT TTC CCT ACA CGA CGC TCT TCC GAT C-s-T-3´
5´-/5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTC dU ACACTCTTTCCCTACACGACGCTCTTCCGATC-s-T-3´
The following sequences are used for adaptor trimming of NEBNext adaptors for Illumina: Read 1 AGATCGGAAGAGCACACGTCTGAACTCCAGTCA Read 2 AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT
| PRODUCT | INDEX PRIMER SEQUENCE | EXPECTED INDEX PRIMER SEQUENCE READ |
|---|---|---|
| NEBNext Index 1 Primer for Illumina (10 µM) | 5´- CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGA TC-s-T-3´ | ATCACG |
| NEBNext Index 2 Primer for Illumina (10 µM) | 5´- CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGA TC-s-T-3´ | CGATGT |
Can we use our WGBS custom indeces/adaptors with this kit?
- The index primers we have in lab are listed here (and I downloaded a version 9/15/2024 here)
- these were designed for WGBS with the Zymo Pico Kit
- Each of the 60 pairs has a unique i7 and unique i5 sequence, for example:
| UDIndexName | i7_ | i7Bases (Sample Sheet) | i5Bases (sample sheet) | i7_ | i7_UDI Sequence | i5 | i5_UDI |
|---|---|---|---|---|---|---|---|
| UDI0001 | AACCGCGG | CCGCGGTT | AGCGCTAG | i7_UDI0001 | CAAGCAGAAGACGGCATACGAGATAACCGCGGGTGACTGGAGTTCAGACGTGT | i5UDI0001 | AATGATACGGCGACCACCGAGATCTACACAGCGCTAGACACTCTTTCCCTACACGAC |
| UDI0002 | GGTTATAA | TTATAACC | GATATCGA | i7_UDI0002 | CAAGCAGAAGACGGCATACGAGATGGTTATAAGTGACTGGAGTTCAGACGTGT | i5UDI0002 | AATGATACGGCGACCACCGAGATCTACACGATATCGAACACTCTTTCCCTACACGAC |
- the i7_UDI Sequence contains:
- CAAGCAGAAGACGGCATACGAGAT
- Illumina P7 adaptor: 5’- CAAGCAGAAGACGGCATACGAGAT -3’
- T overhang
- Illumina P7 adaptor: 5’- CAAGCAGAAGACGGCATACGAGAT -3’
- unique i7_ sequence (UDI0001: AACCGCGG, UDI0002: GGTTATAA)
- GTGACTGGAGTTCAGACGTGT
- Reverse completment: ACACGTCTGAACTCCAGTCA
- CAAGCAGAAGACGGCATACGAGAT
- the i5_UDI Sequence contains:
- AATGATACGGCGACCACCGAGATCTACAC
- Illumina P5 adaptor: 5’- AATGATACGGCGACCACCGAGATCTACAC -3’
- unique i5_ sequence (UDI0001: AGCGCTAG, UDI0002: GATATCGA)
- ACACTCTTTCCCTACACGAC
- reverse complement: GTCGTGTAGGGAAAGAGTGT
- AATGATACGGCGACCACCGAGATCTACAC
Since these are T-overhang adaptors, with what I believe are universal PCR primers, we can hopefully use these indeces with this kit.
There are no exact instructions on how to modify the protocol with use of custom adapters/primers, but this is how the general NEB RNA library prep protocol is modified for custom adapters/primers:
Original Protocol –> Modified protocol
- 5 uL of the Dual Index adapter is used instead of 2.5 uL of the NEBNext Adaptor
- “Proceed immediately to Purification of the Ligation Reaction.” after the 15 minute incubation instead of adding the USER enzyme continued by another incubation.
- In the bead cleanup, a ratio of 0.7X of beads is used instead of 0.9X. And then, the elution is 20 uL instead of 15 uL.
- In PCR enrichment, no index primers are added at this step (since they are included in the adapters above). The NEBNext primer mix is used instead.
- The primer-dimer and adaptor-dimer peaks will be at different expected bps depending on which adaptor/primer set was used