Extracting RNA and DNA from Fixed P. acuta Sections (newly fixed 2/8/24)
Extracting RNA and DNA from Fixed P. acuta Sections (newly fixed 2/8/24)
Continuation from this post, and this post, in which I was able to get relatively good RNA from sectioned tissue from P. compressa and M. capitata.
02/08/24 - Fixing tissue, decalcifying, and preparing for sectioning
Protocol here
- Make 50mL of 1X PBS in RNAse Free H2O and keep at 4ºC for this step and following extraction steps.
- Fixing two “chunky” branches from a large BT3/4 POC colony in one 5mL container with 5.5mL of Fixative.
- Day 1 (02/08/24): Fixed tissue in PAXgene fixative
- sterilized, RNAse cleaned clippers and forceps used for all steps
- Day 2 (02/09/24): Replaced fixative with stabilizer, transferred to 4 ºC
- Day 3 (02/10/24): Started decalcification, kept on shaker in cold room (4 ºC)
- Day 5 (02/12/24): EDTA changed after 48 hours, tissue still not floating, still bits of skeleton.
- Day 7 (02/14/24): Decalc done after 96 hours. Wanted to stop at 72 hours but campus was closed with storm.
- Still wasn’t floating, but seemed clear and ready to go.
- Tissue washed (10 mins, 1X RNAse-free PBS).
- Transferred one branch into Paxgene tissue stabilizer for 6 hours at 4 ºC
- Leave in PAXgene stabilizer at 4ºC until other branch cryoprotection is complete.
- Transferred one branch into 30% Sucrose in RNAse-free PBS for 6 hours at 4 ºC (see notes below)
- Add 3 g sucrose + 10 mL RNAse Free PBS in a 15-mL Falcon tube, vortex to dissolve
- Perform “cryoprotection” by transfering branch from PAXgene stabilizer to the sucrose solution at 4ºC for several hours or overnight, once the tissue sinks you can continue processing.
- Check every hour to see if it sunk. Record hours in sucrose.
- Day 1 (02/08/24): Fixed tissue in PAXgene fixative
- Embedding
- Embed both branches on afternoon 2/14/24 (6 hours from 8:30AM sucrose/stabilizer step) on powereded dry ice, place in -80 ºC overnight for sectioning in morning.
Notes on plan with cryoprotection
- On 2/1/24, I attempted to section a branch from this series that I embedded on 1/29/24 without sucrose cryoprotection and the sectioning failed. The tissue was melting out of the block of OCT, much like the very first time I sectioned P. acuta, which was also without a sucrose cryoprotection step.
- Therefore, I want to test if I am able to section a newly-fixed branch without cryoprotection (not one that has been sitting in the -80 for months), but at the same time I will have a back-up, cryoprotected branch from the same colony to also test RNA and DNA extractions with if the non-sucrose branch sectioning fails.
- Ideally, I am able to section and maintain morphology without a sucrose step as this significantly increases RNA quality
2/15/24 - Sectioning
- Section, make sure temp is around -19 to -20 ºC and play around with the blade angle as necessary
- Keep slides outside of cryostat so they are warm and allow for cold sections to adhere to the slide
- “Cut sections onto slide, then air dry PEN slide at room temp for 2 minutes on dessicant
- Once slide has been dried for 2 mins, place in 50 mL falcon tube with silica gel packet on dry ice and place in -80 ASAP
- (2 slides back to back per tube)
-
After sectioning, transported slides to Molecular lab and proceeded with RNA extraction within 1 hour
- Notes from sectioning
- the non-sucrose P. acuta branch sectioning failed miserably, like on 2/1. So it wasn’t just the old tissue that was the issue. I think there is something about the alcohol in the PAXgene stabilizer that prevents freezing of the tissue; it becomes gummy and the blade is unable to cut through
- got 3 slides from the sucrose-protected tissue, testing one for RNA and DNA extraction, other two put into -80 with dessicant
2/15/24 - RNA Extraction using protocol modificiations used for P. compressa and M. capitata
Protocol Link
Modifications to protocol:
- Make 100 uL of lysis buffer mixture and pre-cool on ice
- Pre-cool forceps and razor blades for transferring tissue to lysis buffer
- 2 minute in ice-cold 70% Ethanol
- 45 s in ice-cold 50% Ethanol
- 30 s in ice-cold 30% Ethanol
- Remove slide to dark background over PCR rack (over dry ice, though be careful to not freeze the water once the volume on the slide is low, if that’s freezing move to regular ice) and use sterile razor blade, cleaned with RNAse cleaner, to scrape off tissue sections into a tube filled with prepared 100 uL RD2 digestion buffer.
- this is double the normal RD2 amount, because I added a lot of tissue
- honestly just used my tiny forceps this time to scoop up tissue, they were cleaned with ethanol and RNAse cleaner
- Then, I followed the protocol as written for FFPE, with a 15 minute digestion at 56 ºC (shaker-incubator, 1400 rpm)
- since I used double the amount of lysis buffer, I used double the amount of binding buffer (200 uL RB7)
- For 2 samples: 176 ul 100% isopropanol + 44 ul RB7
- makes 210 ul, enough for 2 samples + 10% error
- For 2 samples: 176 ul 100% isopropanol + 44 ul RB7
- since I used double the amount of lysis buffer, I used double the amount of binding buffer (200 uL RB7)
Low input sample: 2 polyps worth of tissue put into digestion buffer
High input sample: 2 sections worth of tissue put into digestion buffer
Qubit Results
- Used Broad range RNA Qubit Protocol
- All samples read twice, standard only read once
RNA Standards: BR: 374.17 (S1) & 9837.62 (S2) HS: 30.93 (S1) & 368.43 (S2)
| sample_id | Species | RNA_QBIT_1 | RNA_QBIT_2 | RNA_QBIT_AVG |
|---|---|---|---|---|
| Low_Input (HS) | Pocillopora acuta | 4.40 | 4.40 | 4.40 |
| High_Input (BR) | Pocillopora acuta | 13.4 | 13.2 | 13.3 |
RNA Quality Check: Tapestation

Full results can be found here
Notes on RNA:
These are both very degraded. And just not a lot of RNA. When I did the successful POR and MON extractions, I used more tissue and I also did the 15 minute incubation in the incubator genie instead of the thermomixer. I will try the incubator genie again.
I guess there is a potential that these were not fixed very well? Two branches in 5mL of fixative. And, I only was able to section the sucrose-protected tissue and we know the sucrose step adds degredation. Maybe there needs to be some dehydration step somewhere?
2/19/24 - RNA Extraction using protocol modificiations used for P. compressa and M. capitata
Protocol Link
Modifications/Notes to protocol:
- same as above
- Scrape off tissue sections into a tube: struggled A LOT with this today, tissue was very stuck on slides, which is great, but it took me a long time to scrape into the tubes
- Then, I followed the protocol as written for FFPE, with 3 different versions of the digestion step:
- since I used double the amount of lysis buffer, I used double the amount of binding buffer (200 uL RB7)
- For 3 samples: 528 ul 100% isopropanol + 132 ul RB7
- makes 660 ul, enough for 3 samples + 10% error
- For 3 samples: 528 ul 100% isopropanol + 132 ul RB7
- since I used double the amount of lysis buffer, I used double the amount of binding buffer (200 uL RB7)
Samples:
- Shake: 15 minute digestion at 56 ºC (shaker-incubator, 1400 rpm)
- No shake: 15 minute digestion at 56 ºC (shaker-incubator, 0 rpm)
- Incubator Genie: 15 minute digestion at 56 ºC (incubator genie, shaking speed 25)
Qubit Results
- Used High Sensitivity RNA Qubit Protocol
- All samples read twice, standard only read once
RNA Standards: HS: 30.78 (S1) & 360.61 (S2)
| sample_id | Species | RNA_QBIT_1 | RNA_QBIT_2 | RNA_QBIT_AVG |
|---|---|---|---|---|
| Shake | Pocillopora acuta | nd (0) | nd (0) | nd (0) |
| No shake | Pocillopora acuta | nd (0) | nd (0) | nd (0) |
| Incubator genie | Pocillopora acuta | nd (0) | nd (0) | nd (0) |
Notes: Hmm! this feels wrong. I want to try this again to really hone in on if the incubation is the source of degredation.
2/19/24 - RNA Extraction using protocol modificiations used for P. compressa and M. capitata
Protocol Link
Modifications/Notes to protocol:
- Trying the protocol from this morning again with the following modifications to attempt to reduce tissue loss:
- washing slide on dry ice in 100% ethanol for 30s-1min only, and then going straight into the dissection phase.
- Also using a different p200.
- Then, I followed the protocol as written for FFPE, with 3 different versions of the digestion step:
- since I used double the amount of lysis buffer, I used double the amount of binding buffer (200 uL RB7)
- For 3 samples: 624 ul 100% isopropanol + 156 ul RB7
- makes 780 ul, enough for 3 samples + 50% error
- For 3 samples: 624 ul 100% isopropanol + 156 ul RB7
- since I used double the amount of lysis buffer, I used double the amount of binding buffer (200 uL RB7)
Samples:
- Shake: 15 minute digestion at 56 ºC (shaker-incubator, 1400 rpm)
- No shake: 15 minute digestion at 56 ºC (shaker-incubator, 0 rpm)
- Incubator Genie: 15 minute digestion at 56 ºC (incubator genie, shaking speed 25)
Qubit Results
- Used High Sensitivity RNA Qubit Protocol
- All samples read twice, standard only read once
RNA Standards: HS: 30.07 (S1) & 341.39 (S2)
| sample_id | Species | RNA_QBIT_1 | RNA_QBIT_2 | RNA_QBIT_AVG |
|---|---|---|---|---|
| Shake | Pocillopora acuta | 9.42 | 9.50 | 9.46 |
| No shake | Pocillopora acuta | 7.72 | 7.78 | 7.74 |
| Incubator genie | Pocillopora acuta | 6.52 | 6.58 | 6.55 |
Yay!
RNA Quality Check: Tapestation
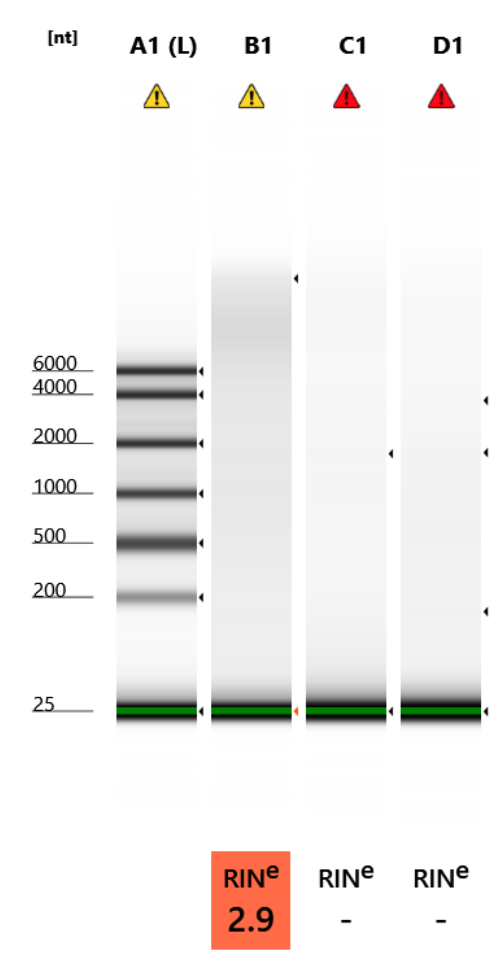

Full results can be found here
Hmm. Tapestation looks worse than the 15th. But, good to see the concentrations up. Incubator genie, though faint, seems to have the best 18S/28S peaks. Might be best to reduce degredation but the concentration is So Low! Could try to increase time of incubation using incubator genie?
2/16/24 - DNA Extraction, protocol here
Same slide and tissue amounts as above, followed protocol as written, very similar to this post for POR and MON
- Followed steps for 3 hr incubation, 60 ºC in thermomixer, no shaking
- pipetted up and down once during the 3 hour incubation to mix
Low input sample: 2 polyps worth of tissue put into digestion buffer
High input sample: 2 sections worth of tissue put into digestion buffer
DNA Standards: BR: 217.27 (S1) & 22522.93 (S2)
| sample_id | Species | RNA_QBIT_1 | RNA_QBIT_2 | RNA_QBIT_AVG |
|---|---|---|---|---|
| Low_Input | Pocillopora acuta | 2 | 2 | 2 |
| High_Input | Pocillopora acuta | 2.82 | 2.74 | 2.78 |
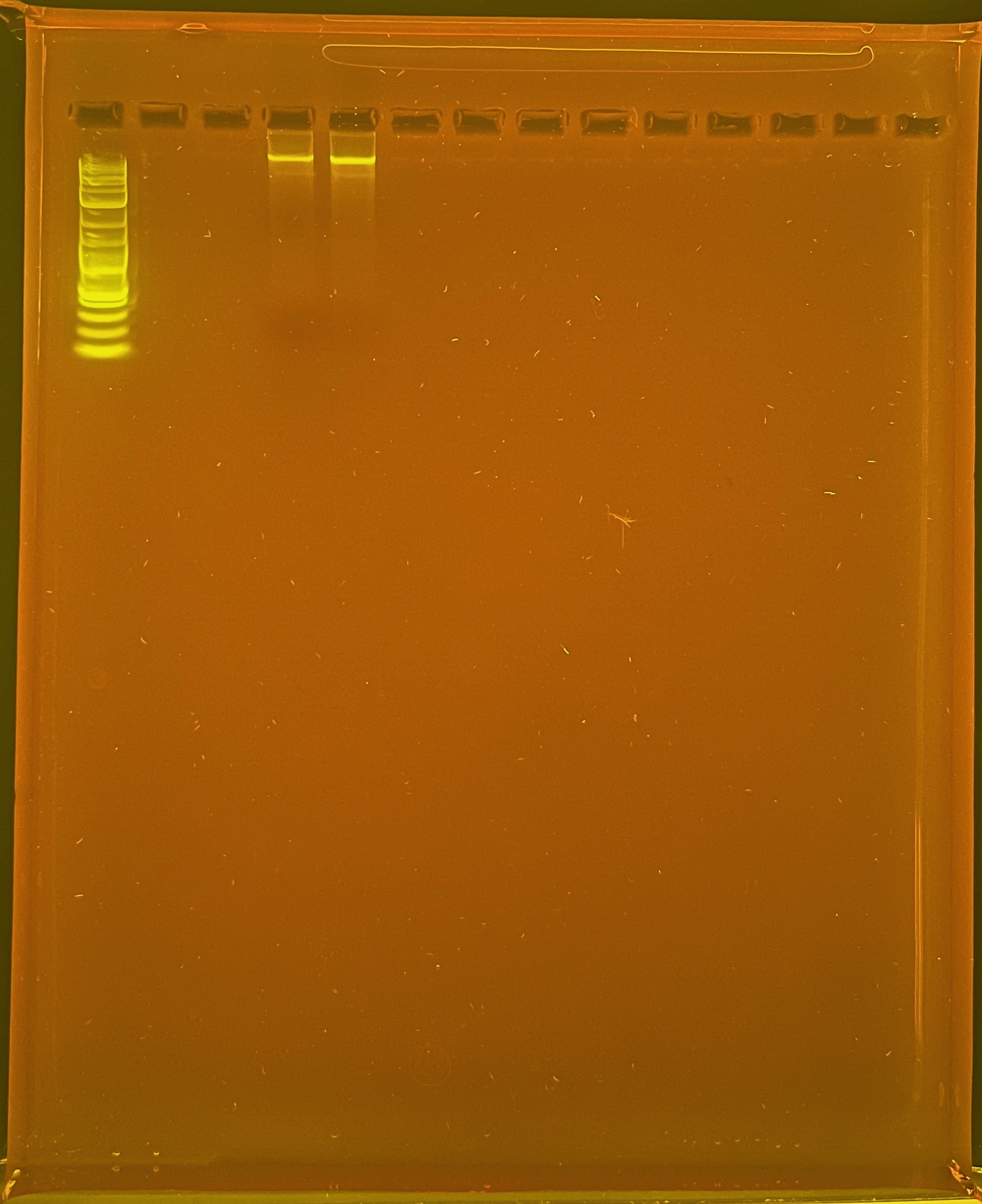
DNA Quality
PPP Agarose Gel Protocol to determine DNA quality. “Good” DNA should form a distinct band a the very top of the gel.
Ran gel next day: